|

CGMP制药洁净室---
细胞治疗产品、单克隆抗体、疫苗以及口服固体抗病毒药物的生产
目前,全球仍在继续竞相寻找抗击COVID-19 爆发流行的最有效策略。迫切需要有效的公共卫生干预措施和可靠的治疗手段来扭转这一激增趋势,与此同时也推动了多种用于改善患者预后的药物开发。
cGMP生产设施
在药物开发和制造的所有阶段,必须遵守国际和国家法规,这保证了这些无菌产品在投放市场时的安全性和有效性。
当前良好生产规范(cGMP)是一项由食品和药物管理局(FDA)、世界卫生组织 (WHO)、欧洲药品管理局(EMA)、药品检验联合检验计划(PIC/S)、治疗品管理局(TGA)和其他地方监管机构等执行的法规。这种正式的法规在充分实施后,可以避免造成污染、混淆、偏差、故障和错误,并确保药品符合其质量标准。
根据FDA cGMP和无菌指南,有两个对无菌药品质量特别重要的清洁区域:
关键区域
该区域被视为关键区域,因为该区域内的已灭菌药品易受污染,且在后续过程中不能对其直接接触的容器进行灭菌。因此,必须对周围环境进行严格控制,以保持产品的完整性和无菌性。
在该区域开展的活动包括灌装区域、产品密封操作之前和期间过程对无菌材料进行操作(例如,无菌传递、无菌材料添加等)。
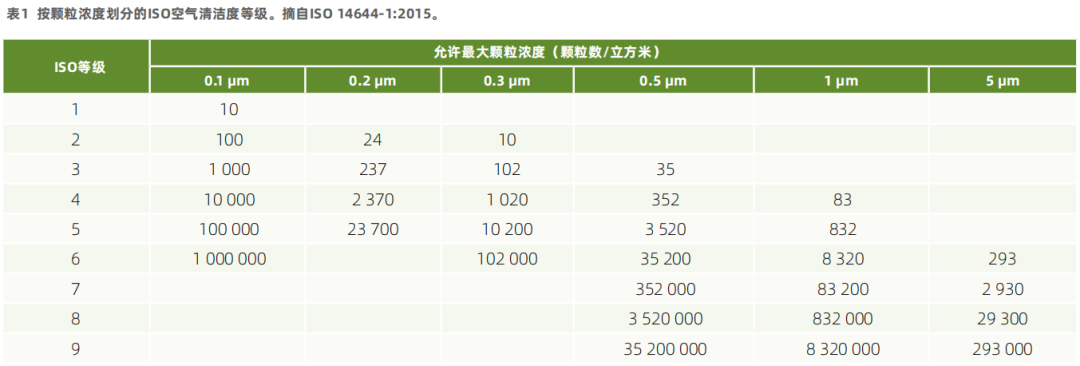
区域中气溶胶颗粒数目非常重要,因为它们可能通过作为微生物载体而对产品造成外来或生物污染。根据FDA的cGMP/无菌指南以及ISO 14644-1:2015,关键区域空气洁净等级为ISO 5级。
ISO 14644-1规定,空气清洁度由每立方米允许的最大颗粒浓度进行分类。
辅助清洁区域
辅助清洁区域可以发挥多种功能。通常,这些区域是准备、操作或转移非无菌成分、产品配方、产品处理中需要的材料、设备、和容器的地方。
辅助洁净区的空气洁净度分类取决于此处开展活动的性质。美国食品和药品监督管理局建议,在动态条件下,无菌处理附近的直接接触区域应符合ISO 7级(最低)动态标准。生产厂家也可以将该区域划分为ISO 6级或将整个无菌灌装室保持在ISO 5级的水平。而按照 ISO 8级空气清洁度等级分类的区域适用于非关键操作(例如,设备清洁等)。
一个区域的ISO分类等级越高,运营成本就越高。由于增加了大量的HEPA和ULPA过滤器,风机和暖通空调的功耗也随之增加,以满足所需的空气洁净等级。
符合cGMP的洁净室
洁净室旨在为无菌和非无菌产品的加工提供高水平的清洁度,以确保产品的质量、安全性和加工工艺效率。
根据操作要求,洁净室环境需要控制单位体积的空气中所含确定数量的颗粒。为了实现这一控制,高效空气过滤器(HEPA)和超高效空气过滤器(ULPA)被投入使用,其目的是用来捕获和限制进入洁净室的颗粒数量。此外,风速也需保持在一定水平[根据相关无菌操作指南,通常为0.45 m/s(90 fpm)±20%],以确保受控空间内每小时有足够数量的换气次数(ACPH)。
上述条件将有助于降低污染风险,同时提供合适的环境来进行相关操作。还可以根据需要控制其他相关物理参数,例如温度、湿度、压缩空气参数、噪音和振动以及静电放电等。
彦雯摘编
版权声明:
本站部分内容、观点、图片、文字、视频来自网络,仅供大家学习和交流,真实性、完整性、及时性本站不作任何保证或承诺。如果本站有涉及侵犯您的版权、著作权、肖像权的内容,请联系我们(021-62511200),我们会立即审核并处理。
|
|||||||||||||||